
Neuro-Ophthalmology Lab
Sun Yat-sen University
State Key Laboratory of Ophthalmology
Sun Yat-sen University
State Key Laboratory of Ophthalmology
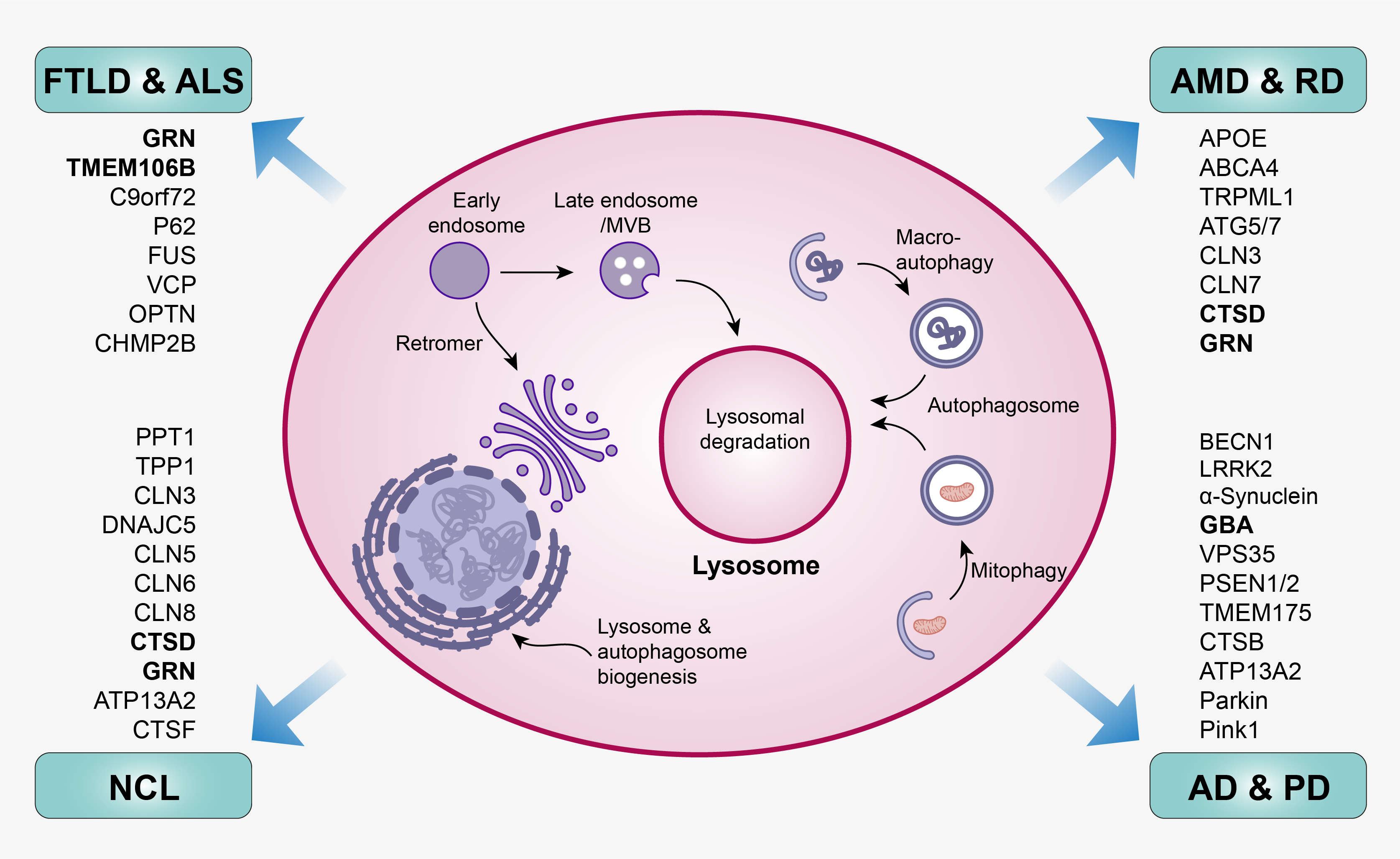
Lysosome is a membrane-bound organelle in mammalian cells. It contains over 60 different digestive hydrolytic enzymes and functions as the “stomach” of the cells to break down unwanted or toxic biomolecules delivered through the endosome-autophagosome-lysosome pathway (EALP). Improper function of lysosomes causes deficits in protein degradation leading to protein accumulation and aggregation as well as triggering neuroinflammation. Accumulating evidence suggests lysosomal dysfunction is a common disease mechanism underlying aging and a variety of neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD), frontotemporal lobar degeneration (FLTD), neuronal ceroid lipofuscinosis (NCL), age-related macular degeneration (AMD) and retinal degeneration (RD). For this theme, our research interest is to 1) study the fundamental mechanism of how lysosomal dysfunction leads to aging and age-related neurodegenerative diseases by focusing on lysosomal basic biology as well as lysosomal-related genes (e.g. GRN, TMEM106B, GBA, CTSD); 2) develop therapeutic treatments against lysosomal dysfunction-related neurodegenerative diseases by screening small drug molecules or applying gene editing to enhance lysosome activities.
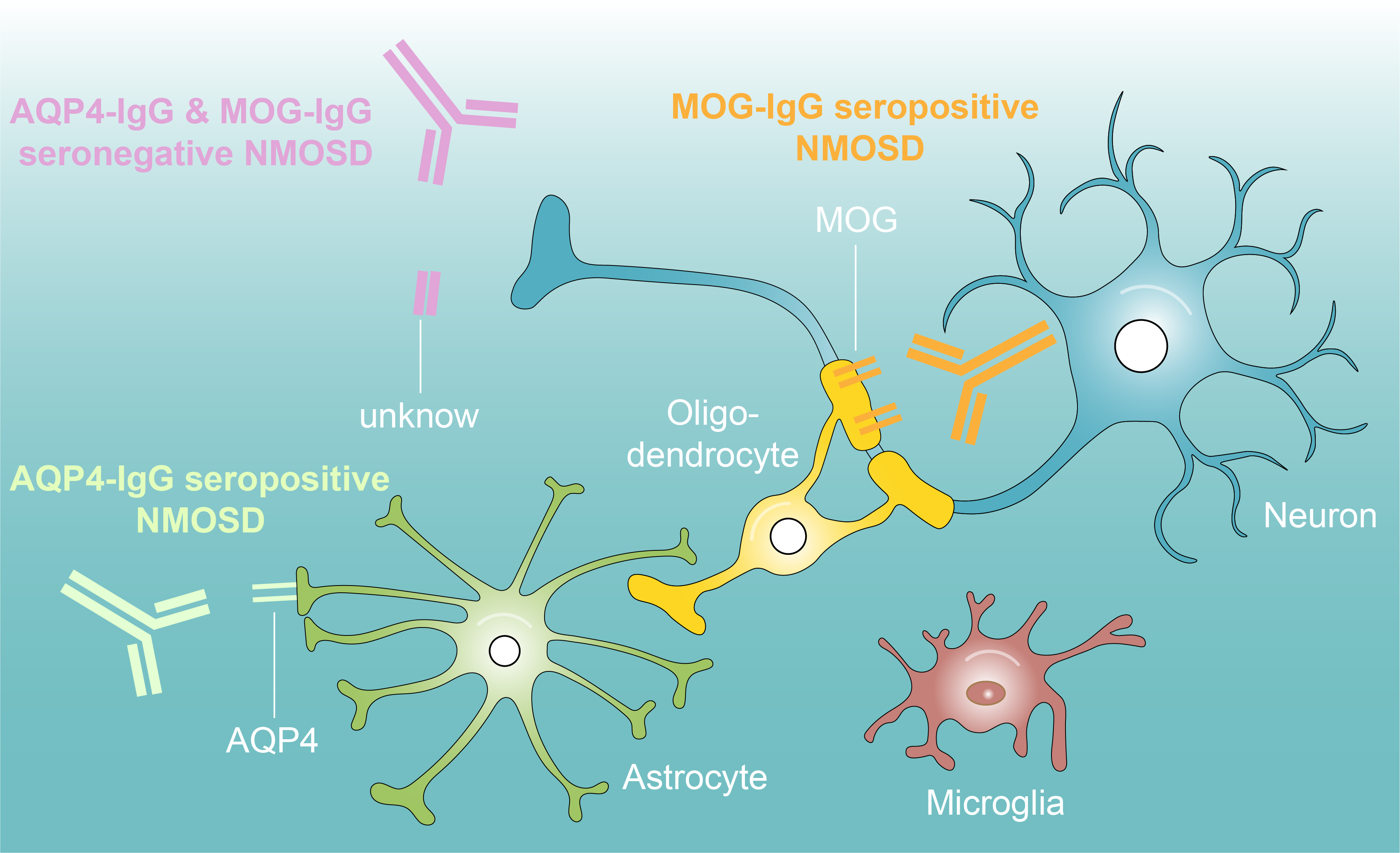
Neuromyelitis optica spectrum disorders (NMOSD), also known as Devic disease, is a devastating auto-immune disease that mainly attacks optic nerve (optic neuritis) and spinal cord (myelitis). Untreated, approximately 50% of patients will be blind and wheelchair users and a third will have died within 5 years of their first attack. However, the detailed disease mechanism is still not fully understood. Pathologically, NMOSD is mediated by auto-immune NMO-associated IgG antibodies, which are reactive to specific structure or functional proteins within nerve tissues and cause demyelination and neuronal degeneration. According to NMO-IgG antibodies, NMOSD is mainly classified into anti-water channel aquaporin 4 IgG antibodies positive NMOSD (AQP4-IgG+, 50-60%), anti-myelin oligodendrocyte glycoprotein IgG antibodies positive NMOSD (MOG-IgG+, 10-15%), and AQP4-IgG and MOG-IgG seronegative NMOSD (AQP4-IgG- & MOG-IgG-, Double negative, 20-30%), for which the pathogenic antibodies remain unknown. Current treatment for acute disease stage includes high-dosage intravenous corticosteroids, intravenous immunoglobulin, and plasmapheresis. Although a majority of the patients (~60%) have a good response to treatments, a marked proportion (~25%) does not respond well. In addition, a significant number of patients will relapse despite the treatment of immune suppression and relapse time is hard to predict. For this theme, we aim to 1) understand the disease mechanisms underlying different NMO-IgG antibodies mediated NMOSD; 2) identify potential novel pathogenic antibodies in AQP4-IgG and MOG-IgG double seronegative NMOSD; 3) identify bio-markers to predict the relapse and prognosis.

Inherited retinal and optic nerve diseases (IRONDs) are the leading cause of blindness in working-age adults. IRONDs is approximately 1 in 2000 individuals, affecting more than two million people worldwide. Over 270 genes listed on RetNet (https://sph.uth.edu/retnet/) have been associated with IRONDs. The most common types of IRONDs include retinitis pigmentosa (RP), cone-rod dystrophy (CRD), Leber congenital amaurosis (LCA), Leber hereditary optic neuropathy (LHON), and dominant optic atrophy (DOA) . Currently, for almost all of them, no cure is available. The clustered regularly interspaced short palindrome repeats (CRISPR)/Cas9 system is a gene-editing technology that can induce double-strand breaks (DSBs). Depending on the presentence of repair template, the repairment of DSBs could generate knockout due to indel mutations or lead to precise editing by using a predesigned repair template. By fusing CRISPR/Cas9 with specific enzymes, the newly developed base editing tools such as cytosine base editors (CBEs) and adenine base editors (ABEs) can achieve C to T and G to A mutations, and prime editor (PE) enables all types of substitutions, transitions, and transversions to be inserted into the target sequence. All these tools offer a great opportunity to treat IRONDs. For this theme, we are interested in 1) screening and optimizing gene editors for different IRONDs by using patient-derived iPSCs and transgenic mouse models; 2) developing deliverable agents for gene therapy.

54 Xianlie Nan Lu
Zhongshan Ophthalmic Center
Research Building 3 Room 901
Guangzhou China 510060
020-87335146
zhouxiaolai@gzzoc.com
粤ICP备2021098991号